
In order to determine if these lava cave bacteria are new species and to understand if they have special genes that allow them to thrive in these extreme environment we first need to “assemble“ the bacterial genome from the DNA we sequence. Imagine the genome as a puzzle; the DNA sequences are the pieces we need to put them together to complete the puzzle.
While this sounds like a daunting task, our aim was to make this process as simple as possible, without the hassle of worrying about software version compatibility and the ever-persistent headache of installing new software. As part of our mission, we aim to make bioinformatics as user friendly as possible with the emphasis that we all have the ability to conduct this type of analyses.
Introducing the Unicellular Long-read Assembly aNd Annotation (Ulana) bioinformatics pipeline. Ulana was developed in-house to provide a fast, convenient, and user-friendly option for assembling and annotating bacterial genomes. The Ulana pipeline can run on any Windows, Mac or Linux based device and is distributed as a docker image; as long as you have docker installed, you can execute the pipeline from any computer. Unfortunately, at this time, Ulana is not compatible with chromebooks or ChromeOS based devices.
Ulana at a glance:
Integrated quality control
De novo assembly and polishing of whole bacterial genomes
Gene annotation
Common genes like: rRNA, tRNA, CYTB
Specialty genes: antibiotic resistant genes, quorum sensing genes (LuxI/LuxR), CRISPR/Cas systems
Tentative species level classification
16S, DNAa, and rpoB BLAST based identification
Important Ulana outputs:
The three most important files that are output by Ulana are 1) the “*_consensus.fasta” file located in the output directory called “medaka” 2) the “*_annotation.tsv” file located in the output directory called “prokkaout” and 3) the “*_report_out.txt” file located in the output directory labeled “ropro_out”.
The “*_consensus.fasta“ file contains the genome assembly which can then be uploaded to TYGS (Type (Strain) Genome Sever) to determine the novelty of the organism. This file can be viewed with any plain text editor (Notepad, Textedit, Notepad++, Atom).
The “*_annotation.tsv” file contains a spreadsheet with the annotations of the genes found within the assembled genome. This file can be viewed and manipulated in any spreadsheet editor (Excel, Google Sheets, Numbers). You can then use the filtering options in any spreadsheet editor to search for specific genes of interest.
The “*_report_out.txt” file contains the tentative identification of the bacteria through BLAST identification of the 16S, rpoB, and DNAa genes. This is a very convenient tool for quickly identifying bacteria to a genus level. This file can be viewed with any plain text editor (Notepad, Textedit, Notepad++, Atom).
Getting started with Ulana
Download and install docker then run the following lines of code in your terminal to download Ulana. In-depth instructions for running Ulana can be found here:
docker pull ethill/ulana:latestPublic repositories for Ulana:
The source code for the pipeline is hosted on github while the current build is hosted on docker hub.
Periodically run “docker pull ethill/ulana:latest“ to update to the latest version.
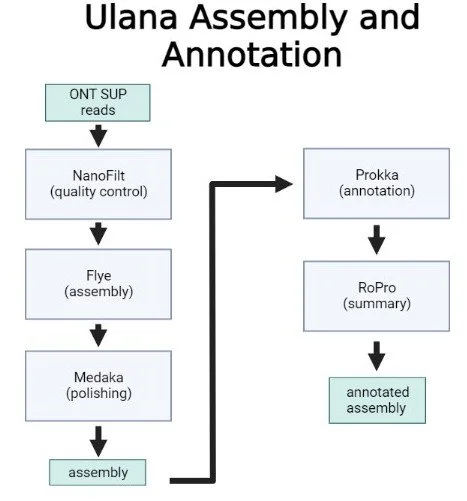
Below is a flowchart of the pipeline that shows each step of the data analysis as well as the tools used to conduct specific analyses within the pipeline.